
Characterization and prevalence of comorbidities in pediatric patients with Down syndrome in the Dominican Republic

 ,
,
Authors
DOI:
https://doi.org/10.37980/im.journal.ggcl.20242324Keywords:
prevalence, comorbilities, Down SyndromeAbstract
This study analyzes the prevalence and characteristics of comorbidities in 534 children with Down Syndrome (DS) in the Dominican Republic, between 2018 and 2022. The research reveals early detection of DS, with an equal gender distribution. Most cases resulted from nondisjunction, with a significant association between advanced maternal age and increased risk of DS. About 62.2% of the children had comorbidities, with cardiac conditions being the most prevalent, followed by endocrine and neurologic comorbidities, mainly hypothyroidism and seizure disorders. Ophthalmic and otorhinolaryngologic conditions were also common, with strabismus and hypoacusis standing out. The findings emphasize the need for early and comprehensive management adapted to the individual and regional characteristics of patients with DS.
Trisomy 21, known as Down syndrome (DS), is characterized by a chromosomal abnormality resulting from the additional presence of chromosome 21, constituting the most frequently observed autosomal aneuploidy compatible with postnatal survival and one of the most complex genetic entities [1]. The estimated global incidence of Down syndrome is approximately 1 per 1000 live births [2].
There are three cytogenetic phenotypes responsible for trisomy 21: (1) free trisomy 21 by nondisjunction, present in 95% of cases and characterized by a total of 47 chromosomes; (2) Robertsonian translocation, with a supplementary chromosome 21 attached to another chromosome, accounting for 4% of cases; and (3) trisomy 21 mosaicism together with partial trisomy 21, which make up 1% of cases and generally correlate with DS manifestations of variable intensity. Excess genetic material induces a number of distinctive clinical manifestations and increases the risk of developing various comorbidities [3].
Several medical conditions are more prevalent in individuals with DS compared to the general population, some of which require immediate interventions after birth, while others warrant continuous monitoring throughout life. It is estimated that approximately 75% of individuals with DS manifest at least one comorbidity of varying degrees of severity [4]. Despite the variability reported in the literature regarding the most prevalent comorbidities in trisomy 21, a preponderance of cardiac anomalies is observed in approximately 44% of cases [5].
It is postulated that the diversity in the types of comorbidities associated with trisomy 21 and their specific manifestations differ significantly worldwide, suggesting an influence of genetic, sociodemographic, and geographic factors [6]. Consequently, various countries have conducted studies to accurately determine the prevalence and variants of comorbidities associated with trisomy 21 in their respective populations.
The prevalence and management of health conditions in individuals with Down syndrome (DS) have been the subject of numerous studies over the years. These studies have provided a deeper understanding of the medical and social needs of this population. According to Chicoine et al [7], individuals with DS in the United States have a high prevalence of common diseases, highlighting the importance of a patient-centered approach to medical care. Lagan et al [8] highlighted the multiorgan involvement and the need for comprehensive management in children with DS, underscoring the diversity of medical needs that require attention.
Previous studies, such as Parker et al [9], have updated prevalence estimates of DS births, highlighting a consistent birth pattern and the need for ongoing resources for these individuals. In line with this, Roizen and Patterson [10] provided a comprehensive review on DS, while Weijerman and de Winter[11] emphasized the importance of clinical practice and specific care for children with DS.
Early mortality and causes of death in individuals with DS have also been the subject of study, as indicated by O'Leary et al [12], who note that comorbid conditions may contribute significantly to morbidity and mortality in this population. In addition, Zigman and Lott [13] explored the relationship between DS and Alzheimer's disease, highlighting the importance of understanding the neurological risks associated with DS.
The epidemiology of DS, discussed by Sherman et al. [14], has provided valuable information on incidence rates and trends. In addition, Korenberg et al [15] investigated DS phenotypes related to chromosomal imbalance, while Gardiner et al [16] discussed the utility of mouse models in DS research. The work of Hassold and Hunt [17] on the genesis of human aneuploidy provides critical insight into meiotic errors leading to DS. Finally, Capone et al [18] addressed the need for a comprehensive approach to the care of adults with DS. These studies collectively provide a solid foundation for understanding the challenges and advances in the care and management of people with Down syndrome.
In the Dominican Republic, there have been unpublished studies focused on congenital cardiac malformations in the pediatric population affected by DS, but there is still a lack of an exhaustive and publicly accessible registry on the comorbidities present in this demographic group. The present research aims to collect and update data on the prevalence of comorbidities linked to trisomy 21 in the Dominican region, with the purpose of detailing each pathology and categorizing them according to their severity or the need for surgical intervention.
An observational, descriptive and cross-sectional research of retrospective nature was implemented, through the analysis of clinical histories of subjects diagnosed with Down Syndrome, who resorted to the medical genetics department of the Dr. Hugo Mendoza Pediatric Hospital, Dominican Republic, covering the interval from August 2018 to August 2022.
The analysis process involved a thorough and systematic review of the existing clinical documentation, without alterations or direct interventions by the researchers on the participants. Subsequently, the data obtained were consolidated in a database designed specifically for this research, using the digital clinical records of the Comprehensive Care Clinic for Patients with Down Syndrome and the Medical Genetics Service of the aforementioned hospital.The methodology for data collection focused on the manual organization of the key information extracted for each patient, using Microsoft Office Excel®. A database structured in five main segments was configured: 1) identification of clinical history, 2) general and sociodemographic information, 3) diagnostic confirmation of trisomy 21, 4) identification of associated comorbidities, and 5) surgical interventions performed, subdividing each category for detailed and accurate cataloguing.Given the observational and retrospective approach of the study, patient participation was not requested, emphasizing the importance of confidentiality and protection of personal data, in line with ethical guidelines.
This procedure ensures the protection of the privacy and integrity of the minors, in strict compliance with the guidelines of the Research Ethics Committee (CEI) of the Hospital Dr. Hugo Mendoza, for exclusively academic and scientific purposes.The initial sample comprised 2,048 patients, from which, after applying the inclusion and exclusion criteria, a final cohort of 534 pediatric subjects with a confirmed diagnosis of Down syndrome was selected. Using probability sampling techniques and establishing a 1% margin of error and a 99% confidence level using Raosoft® software, the representativeness of the sample was validated.
The inclusion criteria defined were: pediatric patients (under 18 years of age) evaluated at the Medical Genetics office of the HPHM in the established period, with a confirmed diagnosis of Down syndrome by genetic analysis. Records with incomplete information, patients seen outside the defined time range and diagnoses based solely on clinical criteria were excluded.
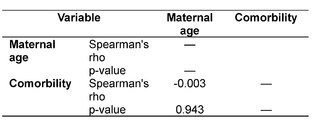
After collecting and categorizing the data, we proceeded to the analysis according to the proposed objectives, applying statistical techniques of frequency distribution, as well as measures of central tendency and dispersion. In addition, JASP® statistical software was used to perform correlations using Spearman's coefficient.
DISCUSION
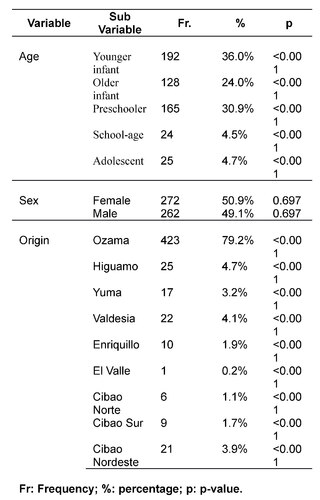
The investigation included 534 pediatric subjects diagnosed with Down syndrome (DS) between August 2018 and August 2022. The gender distribution was nearly equal, with 50.9% (272/534) female and 49.1% (262/534) male participants, evidencing the absence of gender bias in the incidence of DS. The mean age was established at 2.4 years ± 3.1 SD, identifying a higher incidence in the group of younger infants (1-11 months), representing 36% (192/534) of the sample. This data suggests an early detection and approach to comorbidities in DS, potentially contributing to a decrease in morbidity and mortality in this population.
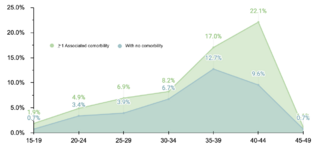
Etiologic evaluation revealed that the majority of DS cases (99%, 529/534) were due to nondisjunction, followed by Robertsonian translocation (0.6%, 3/534) and mosaicism (0.4%, 2/534). These findings are consistent with preestablished etiologic distributions. The mean maternal age at delivery was 34.8 years ± 6.7 SD, with a higher frequency in the 40-44 years range. This pattern reaffirms the association between older maternal age and an increased risk of DS diagnosis.
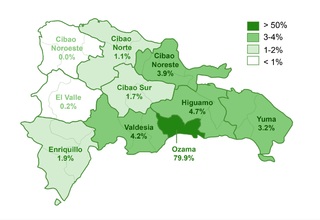
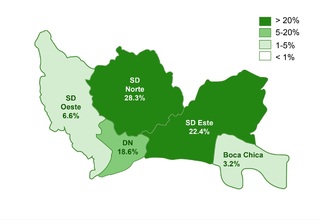
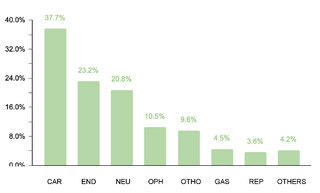
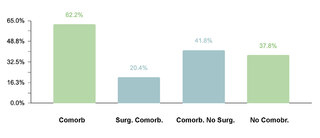
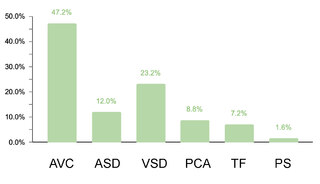
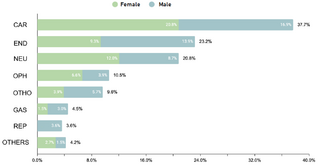
The study identified that 62.2% (332/534) of the sample manifested one or more comorbidities associated with DS, a lower proportion compared to studies conducted in other regions. It was observed that 20.4% (109/332) of patients with comorbidities required surgical interventions. The prevalence of cardiac comorbidities was 37.7% (125/332), with atrioventricular septal defect (AVSD) being the most frequent. Although this percentage is comparable with data from India, it is lower in relation to figures reported in the USA, Spain, Chile and Bangladesh.
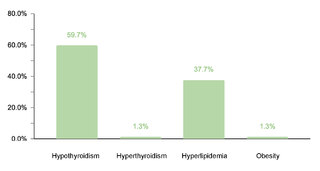
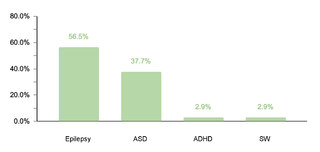
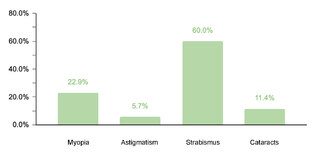
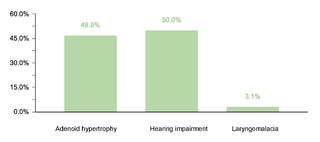
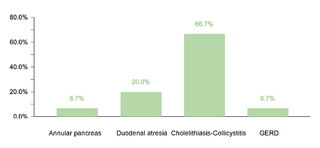
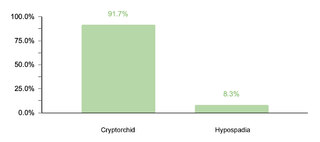
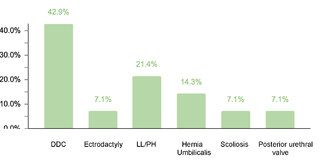
Regarding endocrine comorbidities, 23.2% (77/332) of the patients presented alterations, hypothyroidism being the most common. Regarding neurological comorbidities, 20.8% (69/332) of the cases were diagnosed, with epileptic disorders standing out as the predominant condition. Some 10.5% (35/332) of the children were found to have ophthalmic conditions, mainly strabismus, attributable to the generalized muscular hypotonia typical in DS. Otolaryngologic comorbidities affected 9.6% (32/332) of the sample, with hypoacusis as the most common condition. A smaller percentage showed gastrointestinal and reproductive system comorbidities, with cholelithiasis and cryptorchidism being the most prevalent, respectively.
The research highlights the variability of comorbidities associated with DS and underlines the importance of a comprehensive and early approach to the management of these patients, tailored to the specific needs of each case and based on a thorough understanding of the prevalence and nature of comorbidities in different geographic and demographic contexts.
This study showed that a considerable proportion of the pediatric population with Down syndrome (DS) presents multiple comorbidities, with a particular emphasis on cardiac disorders, highlighting atrioventricular septal defect (AVSD) as the most common. Although gender did not influence the incidence of DS, a specific tendency towards certain reproductive, ophthalmic and gastrointestinal comorbidities was identified as a function of patient gender. In addition, it was reaffirmed that advanced maternal age is a significant risk factor for the development of DS, since a large majority of the cases studied corresponded to mothers older than 35 years.
The need for surgical interventions in a significant portion of patients with comorbidities underscores the complexity of the associated conditions and the importance of specialized medical management. The findings of this study reinforce the need for a personalized medical care strategy for the pediatric population with DS in the Dominican Republic, differentiated from the approaches applied to the general population, in order to improve early detection and treatment of comorbidities, thus reducing associated morbidity and mortality. This approach should be tailored to the specific needs of each patient, taking into account local and demographic particularities to ensure optimal and timely care.
References
[1] Antonarakis SE, Skotko BG, Rafii MS, Strydom A, Pape SE, Bianchi DW, et al. Down syndrome. Nat Rev Dis Primers. 2020 Feb 6;6(1):9. doi: 10.1038/s41572-019-0143-7.
[2] United Nations. World down syndrome day [Internet]. United Nations; 2023 [cited 2023 Feb 9]. Available from: https://www.un.org/en/observances/down-syndrome-day.
[3] Bull MJ. Down Syndrome. In: Ropper AH, editor. N Engl J Med. 2020 Jun 11;382(24):2344–52. doi: 10.1056/NEJMra1706537.
[4] Heinke D, Isenburg JL, Stallings EB, Short TD, Le M, Fisher S, et al. Prevalence of structural birth defects among infants with Down syndrome, 2013–2017: A US population‐based study. Birth Defects Res. 2021 Jan 15;113(2):189–202. doi: 10.1002/bdr2.1854.
[5] Stoll C, Dott B, Alembik Y, Roth MP. Associated congenital anomalies among cases with Down syndrome. Eur J Med Genet. 2015;58(12):674–80. doi: 10.1016/j.ejmg.2015.11.003.
[6] De Rubens Figueroa J, del Pozzo Magaña B, Pablos Hach JL, Calderón Jiménez C, Castrejón Urbina R. Malformaciones cardíacas en los niños con síndrome de Down. Rev Esp Cardiol. 2003;56(9):894–9. doi: 10.1157/13051617.
[7] Chicoine B, Rivelli A, Fitzpatrick V, Chicoine L, Jia G, Rzhetsky A. Prevalence of Common Disease Conditions in a Large Cohort of Individuals With Down Syndrome in the United States. J Patient-Cent Res Rev. 2021 Apr 19;8(2):86–97. doi: 10.17294/2330-0698.1824.
[8] Lagan N, Huggard D, Mc Grane F, Leahy TR, Franklin O, Roche E, et al. Multiorgan involvement and management in children with Down syndrome. Acta Paediatr. Blackwell Publishing Ltd; 2020; 109:1096–111. doi: 10.1111/apa.15153.
[9] Parker SE, Mai CT, Canfield MA, Rickard R, Wang Y, Meyer RE, et al. Updated National Birth Prevalence estimates for selected birth defects in the United States, 2004-2006. Birth Defects Res A Clin Mol Teratol. 2010 Dec;88(12):1008-16. doi: 10.1002/bdra.20735.
[10] Roizen NJ, Patterson D. Down's syndrome. Lancet. 2003 Apr 12;361(9365):1281-9. doi: 10.1016/S0140-6736(03)12987-X.
[11] Weijerman ME, de Winter JP. Clinical practice. The care of children with Down syndrome. Eur J Pediatr. 2010 Dec;169(12):1445-52. doi: 10.1007/s00431-010-1253-0.
[12] O'Leary L, Hughes-McCormack L, Dunn K, Cooper SA. Early death and causes of death of people with Down syndrome: A systematic review. J Appl Res Intellect Disabil. 2018 Jul;31(4):687-708. doi: 10.1111/jar.12446.
[13] Zigman WB, Lott IT. Alzheimer's disease in Down syndrome: Neurobiology and risk. Ment Retard Dev Disabil Res Rev. 2007;13(3):237-46. doi: 10.1002/mrdd.20163
[14] Sherman SL, Allen EG, Bean LH, Freeman SB. Epidemiology of Down syndrome. Ment Retard Dev Disabil Res Rev. 2007;13(3):221-7. doi: 10.1002/mrdd.20157.
[15] Korenberg JR, Chen XN, Schipper R, Sun Z, Gonsky R, Gerwehr S, et al. Down syndrome phenotypes: The consequences of chromosomal imbalance. Proc Natl Acad Sci U S A. 1994 May 24;91(11):4997-5001. doi: 10.1073/pnas.91.11.4997.
[16] Gardiner KJ, Fortna A, Bechtel L, Davisson MT. Mouse models of Down syndrome: How useful can they be? Comparison of the gene content of human chromosome 21 with orthologous mouse genomic regions. Gene. 2003 Mar 6; 318:137-47. doi: 10.1016/S0378-1119(03)00769-8.
[17] Hassold T, Hunt P. To err (meiotically) is human: The genesis of human aneuploidy. Nat Rev Genet. 2001 Apr;2(4):280-91. doi: 10.1038/35066065.
[18] Capone GT, Chicoine BA, Bulova P. Adults with Down Syndrome: A comprehensive approach to care. Curr Opin Psychiatry. 2018 Mar;31(2):113-120. doi: 10.1111/jir.12588
